
Fill in basic information to use the services of NeuSiC
Danish Neuro Single Cell (NeuSiC) platform
The Neuro Single Cell (NeuSiC) platform provides neuroscience groups in Denmark with access to state-of-the-art single cell and spatial omics technologies (10x Genomics, Parse Biosciences, Smart-seq2).
While the user is responsible for the sample preparation (nuclei/cell isolation and potential fixation), NeuSiC covers library generation, sequencing and data analysis. Users are only responsible for covering the expenses for reagents used in library preparation and sequencing, while the working hours are fully subsidized.
The NeuSiC platform is specialized in processing brain tissue from animal models or human samples, as well as other tissues or in vitro models from neuroscience-oriented projects. We are situated at the Biotech Research and Innovation Center (BRIC) in Copenhagen and collaborate closely with BRIC’s Single Cell Core Facility.
The service offered includes all work from library preparation to data analysis, and consists of the following blocks:
- Preparation of single cell/spatial libraries
- Sequencing (Illumina NovaSeq6000)
- Data analysis
User can choose either block 1 only, block 1 and 2, or block 1, 2 and 3.
Users are responsible for nuclei/cell isolation and fixation. Fixation allows for storage of nuclei suspensions and provides a more practical experimental workflow. This option is available only when using the 10x Genomics Flex and Parse kits, but not with the 10x Genomics Universal 3' or 5' kit. For the latter nuclei/cells must be isolated and delivered to our facility on the same day, limiting it to 2–4 samples per day.
Additional information about selecting the appropriate technology is available on the subpage TECHNOLOGIES.
Offered technologies
Single cell omics
- 10x Genomics Universal 3’ Gene Expression (no sample fixation)
- 10x Genomics Universal 5’ Gene Expression (no sample fixation)
- 10x Genomics Flex Gene Expression with sample fixation and multiplexing of samples (only human or mouse samples)
- 10x Genomics Epi Multiome ATAC + Gene Expression (tentatively available in fall 2025)
- Parse EvercodeTM WT Mini (up to 12 samples), EvercodeTM WT (up to 48 samples), and EvercodeTM WT Mega (up to 96 samples) with sample fixation
- Smart-seq2 (full-length transcript coverage) (tentatively available in fall 2025): example
- Patch-seq (tentatively available fall 2025): original paper 1 and paper 2
Spatial omics
- 10x Genomics Visium Spatial Gene Expression (tentatively available in fall 2025)
- 10x Genomics Visium HD Spatial Gene Expression (tentatively available in fall 2025)
Data analysis
There will be two rounds of data analysis. In the initial round, all data from standard pipelines is delivered (specific requests can be discussed in an initial meeting), including an interactive application for the user to assist with further data analysis (example on youtube).
In the final round user-specific adjustments are made and publication-ready figures are delivered.
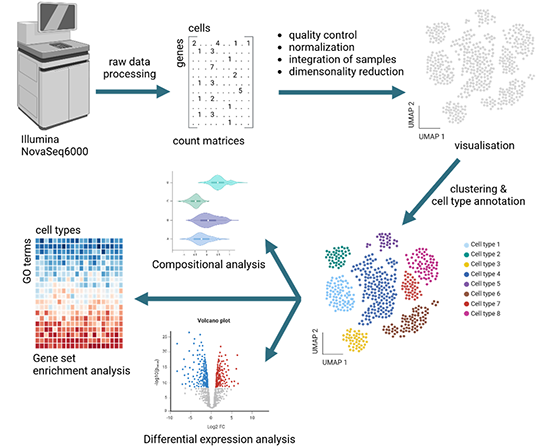
Overview of analysis steps for single cell transcriptomics data.
The raw sequencing data is transformed into count matrices. After quality control and normalization, the samples are integrated and visualized in a UMAP graph. This is followed by cluster estimation and cell type annotation. Standard downstream analysis includes compositional analysis, differential gene expression analysis and gene set enrichment analysis. Additional analyses are available upon request. Figure created with biorender.com.
Standard analysis pipeline for small-scale single cell omics
See also our github repository for example code and plots: https://github.com/NeuSiC/scRNAseq_R_code
1) Raw data processing
- generate fastq files
- produce count matrices
- initial quality control of sequencing and cell calling
2) Quality control of cells (CRMetrics)
- remove low-quality cells based on UMI counts (example) and mitochondrial read fraction (example)
- remove potential doublets (example)
- optional: CellBender to remove ambient RNA (example)
3) Initial data analysis (conos)
- normalization, pre-processing (pagoda2)
- integration of different samples/conditions
- dimensionality reduction
- clustering
4) Cell type annotation
- automated cell type annotation
- or optional performed by user
5) Differential analysis (cacoa):
- Compositional changes (example)
- Transcriptomic expression shifts (example)
- Inspection of sample differences (example)
- Differentially expressed gene analysis (example)
- Genes set enrichment analysis (example)
Additional analysis (user-specific)
Users are only responsible for covering the expenses for reagents used in library preparation and sequencing. If data analysis is requested, a data storage fee of 82 DKK per Tb per month is applied. Our working hours are fully subsidized.
|
10x Genomics Universal 3’ Gene Expression (v3.1) |
4 samples |
8 samples |
16 samples |
|
Reagents and consumables |
50.476 DKK |
100.952 DKK |
201.904 DKK |
|
Sequencing 20.000 reads/cell x 10.000 cells (kit + machine time) |
23.400 DKK |
39.600 DKK |
69.300 DKK |
|
10x Genomics Flex Gene Expression |
4 samples |
16 samples |
64 samples |
256 samples |
|
Reagents and consumables (including fixation) |
56.500 DKK |
138.000 DKK |
313.000 DKK |
830.000 DKK |
|
Sequencing 10.000 reads/cell x 10.000 cells (kit + machine time) |
23.400 DKK |
39.600 DKK |
114.000 DKK |
342.000 DKK |
|
Parse Evercode |
WT Mini kit |
WT kit |
WT Mega kit |
|
cells/nuclei per kit |
10.000 - 20.000 (up to 12 samples) |
100.000 (up to 48 samples) |
1.000.000 (up to 96 samples) |
|
Reagents and consumables (without fixation*) |
35.300 DKK |
79.400 DKK |
141.500 DKK |
|
Sequencing 30.000 reads/cell (kit + machine time) |
23.400 DKK |
69.300 DKK |
342.000 DKK |
*Price of reagents for fixation of cells or nuclei depends on number of samples to be loaded on Parse kit, should be estimated separately (fixation of 1 sample: 300 DKK).
In single-cell transcriptomics, careful experimental design is crucial to minimize batch effects and confounding factors that could lead to spurious clustering and mask the true biological signal. Ensuring an adequate number of samples and incorporating appropriate controls tailored to your research question is equally important to generate robust and interpretable data.
This guide provides key considerations for designing your single-cell experiment. We do not take responsibility for the completeness of the information provided. Always adapt your experimental design to the specifics of your study.
What is a batch effect?
A batch effect refers to systematic differences in your data caused by technical factors rather than biological variation. These effects can arise due to:
- Differences in sample preparation protocols
- Optimize your sample preparation protocol and maintain consistency! Always use the same reagents and equipment.
- The person performing the sample preparation
- Ideally, a single person should handle all samples to minimize variability.
- The day of sample preparation
- Process as many samples as possible per day to reduce variability across batches.
Some batch effects are unavoidable, such as those caused by processing samples on different days or involving multiple individuals in sample preparation. To mitigate their impact, it is essential to:
- Record all batch-related variables (e.g. date, operator, reagent lot numbers).
- Ensure a balanced experimental design, distributing conditions (e.g. treatment, genotype) and biological covariates (e.g. sex, age, post-mortem interval) evenly across batches.
- Address batch effects during data analysis if necessary.
How to design your single-cell experiment:
- Define sample groups and controls
- Determine the number of samples per group based on your research question.
- Ensure appropriate controls (e.g., age-matched samples).
- Identify key biological covariates
- Common covariates include sex, age, treatment, genotype, and post-mortem interval (for human samples).
- Plan sample preparation to minimize batch effects
- Distribute covariates evenly across batches.
- Avoid processing only control samples on one day or all younger samples first.
- Create a sample processing schedule and document it for downstream analysis.
By carefully planning and recording your experiment, you can reduce batch effects and improve the reliability of your single-cell data.
Our in-house protocols for nuclei preparation
- Nuclei isolation with gradient
- Protocol for nuclei isolation without fixation: NO_FIXATION_Nuclei_isolation_with_gradient.pdf
- Protocol for nuclei isolation for subsequent fixation for 10x Genomics Flex or Parse Biosciences: FOR_FIXATION_Nuclei_isolation_with_gradient.pdf
Protocols for nuclei/cell fixation are available on the companies webpages
- 10x Genomics FLEX
- Parse Biosciences:
Contact us
Mail: neusic@bric.ku.dk
Address
BRIC - Biotech Research & Innovation Centre
Ole Maaløes Vej 5
DK-2200 Copenhagen
Khodosevich group office space (building 3, 4th floor)
Funding
NeuSiC is funded by the Lundbeck Foundation.
We ask that users acknowledge the NeuSiC facility in all scientific publications, presentations, posters or any other public announcement that reference data generated here.
Advisory board
We receive support from an advisory board consisting of:
- Prof. Ana Pombo, MDC, Berlin/John Hopkins, USA
- Prof. Igor Adameyko, Medical University of Vienna, Austria
- Prof. Goran Karlsson, Lund University, Sweden
- Assoc. Prof. Jean-Francois Perrier, University of Copenhagen, Denmark
- Jan Egebjerg, Lundbeckfonden, Senior Vice President, Grants & Prizes, Director of Science
People
Konstantin Khodosevich
Professor, Scientific Head of Facility
Irina Korshunova
PhD, Manager of Facility
Laura Wolbeck
PhD, Computational staff scientist
Eman Ahmad Mouhammad
Wet lab staff scientist